Hu Yunfu: Strategies and Trends of Drug Clinical Companion Diagnosis in New Drug Development
May 28, 2021
In the past few years, the proportion of targeted drugs in the new drug projects approved by the US FDA has gradually increased, from the common EGFR to the rare NTRK. Dr. Hu Yunfu said: "Companion diagnosis is for drugs with'deficiencies'. When a drug only works in a smaller group, companion diagnosis is the key to opening the door for the drug to market."
Hu Yunfu, Chief Medical Officer of Fanshengzi
Companion Diagnostics (CDx) provides essential information for the safe and effective use of related drugs through the detection of Biomarker.
Why do you need companion diagnosis in the development of new drugs, and which drugs need to be accompanied by diagnosis?
In the clinical practice of new drugs, companion diagnosis can accurately target the target population and indications by stratifying or enriching patients, helping the drug to obtain more ideal clinical results. From the perspective of R&D cycle, clinical scale or clinical success rate, companion diagnostics can bring significant benefits to drug R&D units. Therefore, in Dr. Hu Yunfu's view, it would be unwise for pharmaceutical companies to spend hundreds of Million to develop new drugs, but not to spend a few Millions for companion diagnosis. Taking different application scenarios as examples, Dr. Hu Yunfu explained several types of new drug development types that are very necessary to consider for companion diagnosis. For example, when the efficacy of a new drug is not sufficiently different from that of the comparison drug, the assistance of companion diagnostics will be very important to improve the clinical success rate; when the benefiting patients are relatively low in the population, such as some rare fusion studies, the patients are not included in the group. If screening is carried out, it is difficult for clinical trials to succeed. In particular, it should be noted that when the proportion of benefiting patients in the population is very low, the specificity of the detection method has a great impact on the clinical ORR. The specificity of 99% and 98% may sound high, but in fact it may not be sufficient. To give a simple example: assuming that the proportion of Biomarker-positive patients in the patient population is 1%, if the specificity of the test reagent is 99%, it means that on average, there is about 1 false positive in every 2 enrolled patients. The effect will be reduced to 1/2; if the specificity is reduced to 98%, there will be about 2 false positives in every 3 enrolled patients, and the clinical effect of the drug will be reduced to 1/3*. Therefore, it is extremely important for pharmaceutical companies to select companion diagnostics with high accuracy.
Technology Trends of Companion Diagnosis
How to choose a technology platform if you want to perform companion diagnosis in a new drug clinic? Combining technology trends, Dr. Hu Yunfu said that in the FDA’s approval milestones over the past ten years, we can see that the most important changes in companion diagnostics have three characteristics: First, as scientific research and academics continue to deepen their understanding of Biomarker, Continuously driving the clinical demand for Biomarker, the demand for Biomarker detection is iterating from a single target to a composite target. During the work of the FDA, one of the regulatory innovations led by Dr. Hu's team that had a huge impact on companion diagnosis was the definition of specific mutation sites from the past to the current definition of gene classification rules. The second is the continuous expansion of sample types, mainly tissue samples, to the emergence of liquid biopsy sample types, sample selection is more diversified; the third is the iteration of detection technology, from the past to single target qPCR, IHC, FISH Mainly, to the presentation of various types of NGSpanel characterized by Biomarker enrichment. The enrichment features of NGS-based CDx on the one hand can make the choice of Biomarker more flexible for new drug development, and on the other hand, it will further promote the clinical accessibility of drugs after the market. How to choose the technology platform of companion diagnostics for new drug research and development? Dr. Hu Yunfu pointed out that each detection technology has its limitations. When selecting, it should consider factors such as product performance, sample accessibility, registration strategy, clinical application accessibility, and input-output ratio, combined with specific Biomarker Characteristics to make the final choice. For example, in terms of sample accessibility, the current sample size of a patient shows a decreasing trend; in the future, as the window for cancer diagnosis and treatment moves forward, the sample size that can be obtained will further decrease. Under this trend, drugs need to consider whether their companion diagnostic platform can use a small number of samples to compare with more Biomarker for detection in the future. From the characteristics of Biomarker, taking fusion gene detection as an example, there are many different detection methods, but from the comparative data of Genetron Health, we can see that compared with FISH, RT-PCR, DNA-NGS and other methods, it is based on RNA -NGS fusion detection is more sensitive.
What are the pathways of companion diagnosis for new drugs in clinical practice, and what key points should be paid attention to?
The CDx development path for new drug R&D is divided into two categories. The most ideal way is joint development. CDx development is carried out at the same time as drug R&D. The CDx strategy needs to be determined in the early stage of drug R&D clinical trials (recommended phase I). However, in practice, it is often too late or not planned to develop CDx during drug development. The clinical trials of drugs use services provided by clinical testing institutions (CTA, ClinicalTrialAssay). In this case, bridging tests are required to develop CDx. As shown in the figure below, the clinical efficacy of drugs in CTA-positive patients has been known in clinical medicine, and the bridging test needs to determine the efficacy of drugs in CDx-positive patients (that is, the group of patients who will use the drug after the CDx kit is used in the future). Among them, CTA-CDx+ patients are not included in the clinical drug group, so statistical methods need to be used for estimation.
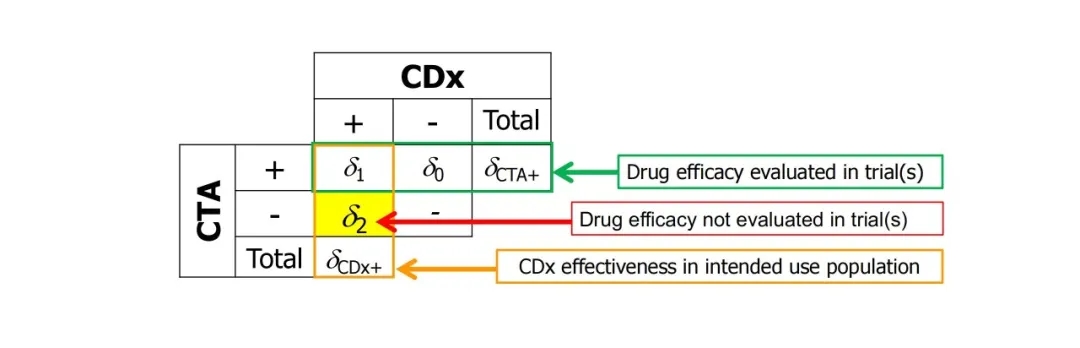
How to estimate? The simplest method is to use CDx to retest the CTA+ and CTA- samples, supplemented by some assumptions and then calculate by weighted average (see the following formula [1]). This is the method used by many FDA-approved companion diagnostics. In actual operation, CTA samples are often not 100% retestable. At this time, special attention should be paid to issues such as the Covariant of testable and untestable samples, and the representativeness of external supplementary samples.

In general, for projects that cannot be co-developed by CDx during drug development and need to bridge trials in the future, Dr. Yunfu Hu recommends the following planning: When drug clinical trials are carried out, it is best to choose CTA with in-depth research for the key Enrollment of the trial; ensure the informed consent signed by the patients to allow retesting; store as many positive and negative samples as possible reasonably; finally, potential inconsistencies, data loss, deviations, etc. need to be considered in the statistical analysis planning.
Clinical compliance and Sino-US dual reporting strategy
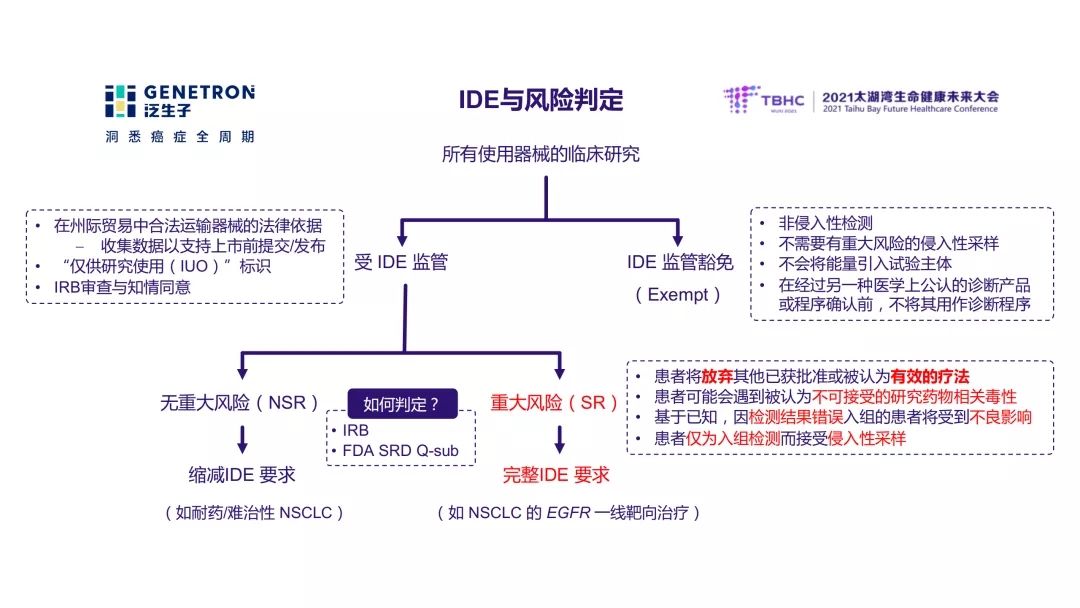
Most clinical trials in the United States are monitored by IDE (Investigational Device Exemption), but not all clinical trials require IDE submission to the US FDA. Dr. Yunfu Hu introduced that only when the device used in the clinical trial is judged to pose significant risk (SR, significant risk) to the patient, the IDE needs to be submitted to the FDA before the start of the clinical trial; there is no significant risk (NSR, Nonsignificantrisk) study There is no need to submit an IDE to the FDA, but it is also monitored by the IDE. How to determine the risk? A typical major risk situation is that patients enrolled in the study need to abandon therapies that have been proven effective. Clinical research units can also communicate with the FDA in advance through Q-sub.
In response to the Sino-U.S. dual reporting strategy focused on new drug research and development, Dr. Hu gave a professional solution: Based on the management content of human genetic resources in my country and the requirements for the scope of use, the simplest and ideal path for Sino-U.S. companion diagnostic dual reporting, It is to choose a diagnostic company with a Sino-US dual center laboratory as a partner to use the same kit to conduct simultaneous Sino-US tests and parallel registration and application on both sides. In this case, foreign data can be merged into domestic analysis. For projects that use two different kits and require a bridging test (which is also a model that is often involved at present) for companion diagnostic reporting, it is still necessary to complete the two kits in clinical trials through cooperators who have the capability of both China and the United States. The test and efficacy data are compared, and the declaration is completed in the two places respectively. In this case, the remaining foreign samples must be combined for domestic testing.

Finally, Dr. Hu Yunfu also introduced the policy trends of similar companion diagnostics in China and the United States and the challenges facing the current CDx development paradigm: including how to submit an IDE if multiple LDTs are used for registration? How to choose LDT negative samples? How to expand the scope of indications after approval, etc. The key issues facing the development of clinical companion diagnostics for these new drugs need to be discussed in depth by pharmaceutical companies, regulators, and diagnostic companies in order to jointly explore a path to companion diagnostics that meets China’s national conditions and is universal. Many excellent pharmaceutical company partners work together to help new drug research and development, promote the entire precision medicine process, and bring more and earlier benefits to clinical patients. The future is here, just around the corner!*It is assumed that the sensitivity is 100%, the ORR of positive patients is 100%, and the ORR of negative patients is 0.
references:
1.LiM.(2015).Statisticalconsiderationandchallengesinbridgingstudyofpersonalizedmedicine.Journalofbiopharmaceuticalstatistics,25(3),397–407.https://doi.org/10.1080/10543406.2014.920340